
What are Inherited Retinal Diseases (IRDs)?
Inherited retinal diseases—or IRDs—are a group of diseases that can cause severe vision loss or even blindness. Each IRD is caused by at least one gene that is not working as it should. IRDs can affect individuals of all ages, can progress at different rates, and are rare. However, many are degenerative, which means that the symptoms of the disease will get worse over time.
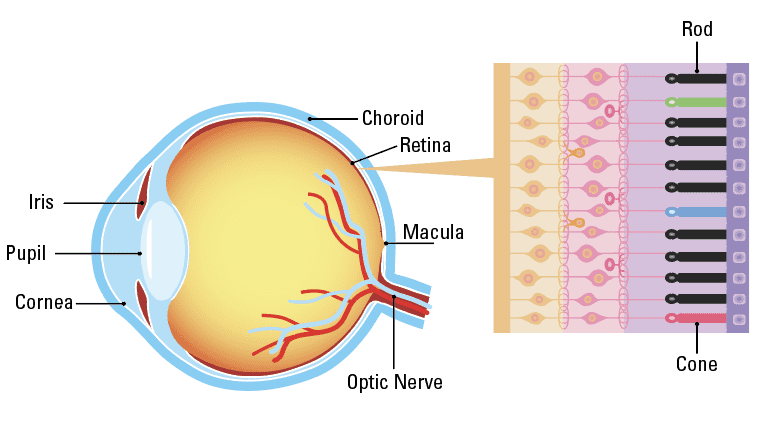
Parts of the Eye
Fact Sheets to Share and Print
There are many types of IRDs identified and others yet to be discovered. The most common types of IRDs include:
Retinitis Pigmentosa (RP)
Retinitis pigmentosa is a group of related eye disorders caused by variations in 60 genes that affect the retina (terms highlighted in teal are labeled on the diagram of the eye above). In people with RP, vision loss occurs as the light-sensing cells of the retina gradually die off. The severity and how fast the disease progresses can vary from person to person with RP, depending on the gene affected. RP can first appear during childhood (early onset RP) or during adulthood. The first sign of RP is usually loss of night vision, called night blindness. Later, RP causes blind spots to develop in the peripheral (side) vision. Over time, these blind spots progress to reduced peripheral vision. The disease progresses over time to eventually affect central vision, necessary for tasks such as reading, driving, and recognizing faces.
Choroideremia
Choroideremia is a condition with progressive vision loss, mostly affecting males. The first symptom of this condition is usually night blindness, which can occur in early childhood. Over time, a person will develop tunnel vision and lose the ability to see details. These vision problems are due to an ongoing loss of cells in the retina and the nearby network of blood vessels (called the choroid). The vision impairment in choroideremia worsens over time, but the rate of worsening varies among affected individuals. This condition may cause complete loss of vision by late adulthood.
Stargardt Disease
Stargardt disease is also called Stargardt macular dystrophy. The disease causes damage to the macula, a small area in the center of the retina that is responsible for sharp, straight-ahead vision. The disease typically causes central vision loss during childhood or adolescence. Sometimes, vision loss may not be noticed until later in adulthood. Only rarely do people with the disease lose all vision.
Cone-rod Dystrophy (CRD)
Cone-rod dystrophy (CRD) is a group of more than 30 IRDs that affect the cones and rods. Cones and rods are the light sensitive cells found in the retina. With progressive deterioration of the cones and rods, people with this condition experience vision loss over time. The first symptoms usually occur in childhood, and may include blurred vision and an intense sensitivity to light (called photophobia). These symptoms are followed by blind spots in the center of vision, loss of the ability to see color, and loss of side or peripheral vision. Most individuals with this condition lose a significant amount of vision by mid-adulthood.
Leber Congenital Amaurosis (LCA)
Leber Congenital Amaurosis is an eye disorder that primarily affects the retina. The retina is the layer of the eye that acts like the film in the camera, capturing a visual image and sending electrical signals to the brain. LCA is one of the earliest onset forms of an IRD. People with this disorder typically have severe visual impairment beginning in infancy. LCA is also associated with other vision problems, such as:
- Photophobia: Increased sensitivity to light
- Nystagmus: Uncontrollable movements of the eyes
- Extreme Farsightedness: An inability to clearly see objects up close, such as a book or watch face.
- Slow reacting pupils: Pupils are the black circles in the center of the eyes surrounded by the iris that change size in response to the amount of light entering the eye. The pupils do not react normally to light for individuals with LCA. Instead, the pupils open and close more slowly than normal, or they may not respond to light at all.
- Misshaped corneas: The cornea is the clear front covering of the eye. It may be cone-shaped and unusually thin with LCA.
- Crossed eye (strabismus): The muscles of the eye do not form or work properly, causing the eyes to look in two different place at the same time.
What are genes?

The human body is composed of trillions of cells. Cells are the basic building blocks of all living things. The command center of each cell is called the nucleus, and it contains chromosomes. Chromosomes are made up of DNA—the body’s hereditary material. A gene is a small section of DNA that contains the instructions for a specific molecule in the body, usually a protein. Each gene contains the information required to build specific proteins needed in the body- proteins build bones, determine eye color, allow muscles to move, control digestion, and keep your heart beating. If there is a change in a gene’s DNA sequence, called a variant, it can cause a necessary protein to not work properly, or to be missing.
What Causes Inherited Retinal Diseases?
An IRD is a genetic disorder– a change, or variant, in one or more genes that contribute to proper retinal function. The genetic disorder affects the gene’s ability to do its job properly. If there is a mistake in a gene, a protein might not be made correctly or at all, and cells in the retina can degenerate and cause vision loss. There are more than 270 different genes known to cause IRDs.
Some gene mutations that cause IRDs are more severe than others. Your doctor is not only interested in which gene is not working properly in a patient, but also how vision is affected. Identifying the specific type of gene variant helps the doctor to provide the correct diagnosis to the patient and may allow him or her to direct patients to clinical trials for therapies that may save their vision. Genetic testing is now available to identify most, but not all, gene variants that cause IRDs.
What Increases the Risk for Inherited Retinal Diseases?
IRDs are diseases that result from variants in our DNA. DNA is the hereditary information provided to a child from its mother and father. Variants in the DNA may be inherited from both parents, one parent, or can occur spontaneously. There are three types of inheritance patterns that can lead to an IRD, including- autosomal dominant, autosomal recessive, and X-linked. It is important to understand the pattern of inheritance to help doctors determine the type of IRD a person may have, and how it might be treated.
Autosomal dominant: A pattern of inheritance in which an affected person receives one copy of a variant dominant gene from one parent and one normal gene from the other parent. The variant dominant gene causes the IRD to occur.
Autosomal recessive: A pattern of inheritance in which the affected individual receives two recessive variant genesone from each parent. The parents are carriers who have only one normal copy of the gene and one variant copy of the gene. The parents do not exhibit the trait because the variant gene is recessive to its normal counterpart gene. If both parents are carriers, there is a 25% chance of a child inheriting both variant genes and developing an IRD. There is a 50% chance of a child inheriting only one variant gene and being a carrier, like their parents. Finally, there is a 25% chance of the child inheriting both normal genes and will not have an IRD or be a carrier.
X-linked disorders: X-linked inheritance means that the genetic variant is located on the X chromosome. These variants can cause X-linked disorders. X-linked variants do not cause the same problems in males and females. X-linked inheritance patterns differ depending on the type of inheritance and variant genes on the X chromosome which can be recessive or dominant. X-linked recessive conditions are always passed on from mother to child, with male children being affected with the condition and female children becoming carriers.
Mitochondrial inheritance: A pattern of inheritance in which the mitochondria (the structures in each cell of the body that are responsible for making energy) are not working properly and do not make enough energy for proper cell function. Mitochondrial inheritance patterns are always passed on from mother to child and can emerge at almost any age. Some mitochondrial genetic disorders can affect one organ, but many of these disorders can affect multiple organs, including the eyes.
Why is Genetic Testing Important?
Identifying the genetic cause of disease is an important part of care for patients with IRDs. Many times, the exact type of IRD a person has can be difficult to determine based only on tests conducted in the eye doctor’s office. Results from genetic testing will lead to an accurate diagnosis. Having the genetic diagnosis will help to identify potential treatment options for patients, inform them about the potential risk of disease to other family members, and identify the potential risk to other organs in the patient’s body that may be affected. In the case of infants and young children, genetic testing will identify those children who are at risk of other health problems and who will benefit from early diagnosis and therapy. The health care provider will order the genetic test, collect the sample, and review the results with the patient. The health care provider may also include a genetic counselor to guide the patient and their family through the results of the genetic testing, discuss the impact on other family members, and guide couples in future family planning decisions.
How are Inherited Retinal Diseases Diagnosed?
When an eye doctor suspects that someone has an IRD, he or she will refer the patient to an ophthalmologist. The ophthalmologist will conduct a very detailed examination including the steps listed below. The goals of the detailed examination are to establish the specific diagnosis, get the treatment that is right for that diagnosis, connect the patient to supportive services such as low-vision rehabilitation, educate other family members about their risk for the disease, and provide information to the patient about clinical trials and new therapies.
The initial and follow-up examinations may include the following:
Patient history: Including current vision issues, patient medical history, current medications, and past medication use.
Family history: Constructing a family medical history to show the genetic relationships and medical disorders that occur in a family. From the family medical history, patterns of familial disorders and how diseases were passed down may emerge. This, in turn, leads doctors to a clear diagnosis of the genetic disorder and allows assessments for family members who may also be at risk for the disorder.
Clinical eye examination: The eye examination includes:
-
- Dilation: Eye drops put into the eye to widen the opening on the front of the eye, called the pupil. This allows the eye doctor to examine the health of the retina
- Visual Acuity: A measure of the ability to see detail up close and in the distance
- Slit lamp: A non-invasive procedure that uses a microscope and bright light to look at different parts of the eye
- Indirect ophthalmoscopy: A non-invasive tool worn on the head of the eye doctor that provides magnification, allowing them to examine the back of the eye
- Imaging: The eye doctor will conduct a series of imaging tests to view different parts of the eye, including:
-
-
- Retinal photographs: A photo taken of the inside surface of the eye
- Optical Coherence Tomography (OCT): A test that takes cross-sectional pictures of the retina. This allows your ophthalmologist to map and measure the thickness of the retina
- Fundus autofluorescence: A test used to create a density map of the layers of the retina
- Infrared autofluorescence: A test that provides information on the distribution of pigment in the retina
- Visual Field Testing: A test that measures central and side or peripheral vision.
- Electroretinography: A test to measure the electrical response of the eye’s light-sensitive cells, called cones and rods. During the test, the eye doctor will place drops into your eyes, so you will not have any discomfort during the test.
- Genetic testing: Patients may meet with a genetic counselor (someone that specializes in the genetic testing process and educating patients) to determine the testing approach that is best based on the other test results. Patients are often asked to provide a blood or saliva sample that will be sent to a lab for analysis. The genetic test confirms or disputes the diagnosis of a specific IRD, so that correct information is provided to the patient and family. Genetic testing is often a requirement for participation in various research trials.
-
The Eye and Gene Therapy
IRDs are strong candidates for gene therapy treatments. The retina is small and easy to access for treatment compared to other parts of the body. Another reason that the eye is an ideal location for gene therapy is that it is considered “immune privileged.” Usually when a foreign substance—like a virus—is detected in our bodies, our immune system works hard to take care of the problem. However, certain areas of the body are immune privileged, which means that our normal immune response isn’t as active. This is typically in areas of our bodies that are very important, and may become damaged if swelling or inflammation occurs. This means that anything that is implanted into the eye—a cell with a corrected gene, for instance—is less likely to be rejected. The most common approach for gene therapy in the eye is delivering the normal gene to the retina using a vector. A vector is a modified virus that will not multiply or cause structural damage. Additional information on gene therapy can be found at: www.asgct.org/education/inherited-retinal-diseases
What therapies are available for Inherited Retinal Diseases?
Not all IRDs have therapies available at this time, however, research is being conducted to develop new therapies that address different types of IRDs. These therapies aim to stop the disease from advancing, to return some degree of sight to patients through targeted therapies, or seek to actively simulate sight through a device called a “retinal prosthetic.”
Types of IRD Clinical Trials:
- Neuroprotective Agents: A neuroprotective agent is a medicine that works to prevent the death of cells in the eye. The therapy designed to slow degeneration of cones and rods, the light sensitive cells in the eye.
- Gene Therapy: Gene therapy replaces a faulty gene or adds a new gene in an attempt to stop, cure disease, or improve your body’s ability to fight a disease. Gene therapy is currently only available for treating IRDs related to a specific gene. [See Sidebar “The Eye and Gene Therapy”]
- Retinal Prosthetic: A retinal prosthetic works to restore vision for patients with certain IRDs by using a microchip that converts images collected by a camera worn by the patient into impulses that are sent wirelessly to the brain.
Patients with an IRD will typically receive therapy (if one is available) and support from many types of medical professionals. These medical professionals provide a team approach to care. This is because IRDs are complicated diseases that can have many impacts on a patient’s life as well as their family. The care team will typically include:
- An IRD specialist: This is often a retina specialist, pediatric ophthalmologist and/or a neuro ophthalmologist
- A genetic counselor
- A low-vision specialist: A professional that provides an evaluation of vision loss and directs patients to tools and methods that will increase independence
- Social workers
- School and education specialists
What Can I Do if I Have an IRD?
Patients with IRDs should play an active role in the treatment of their disease and stay aware of clinical research opportunities. There are a number of tools, services, and resources patients and their families can use to stay informed and find help.
Learn more about IRDs and benefits of genetic testing at eyesongenes.com.
Participate in patient data registries such as My Retina Tracker fightingblindness.org/my-retinatracker- registry. The My Retina Tracker Registry is a research database of people and families affected by IRDs. The registry is designed to share de-identified (i.e. non-traceable to any one individual) information within the IRD research and clinical communities about people with an IRD to help accelerate the discovery of therapies and cures. The registry helps to connect patients with clinical research. There is no charge for the use of this service.
Complete genetic testing when recommended by your doctor. This provides the IRD care team with the most detailed diagnosis and allows them to recommend the best possible treatment for the specific IRD. Genetic testing is also often a requirement before a patient can participate in clinical trials. Find out more about genetic testing, find a provider, or listen to stories from individuals who have completed genetic testing at EyeWant2Know. com. Free genetic testing is now available from select commercial businesses.
Stay up to date on clinical trials at www.clinicaltrials.gov.
Participate in a support group for patients with an IRD, or connect to an online community. These groups are an important way for patients and their families to find support, learn ways to cope with an IRD diagnosis, and help the patient enjoy a better quality of life.
Connect with a local organization that provides services and support for people who have vision loss or are blind. Every state has a department of rehabilitation supported by the National Council of State Agencies for the Blind.
Support services include vocational rehabilitation (including job retraining), mobility training, evaluation for assistive technology devices, and individualized counseling. Local support services can be found at www.ncsab.org or visionservealliance.org/visionloss-resources/lost-your-vision. A variety of organizations supporting individuals with an IRD can be found at: www.asharedvision.com.
Seek out resources to support a high quality of life. Prevent Blindness offers resources and help finding clinical trials for individuals with vision loss on the Living Well with Low Vision website: lowvision. preventblindness.org. Many IRD patients with vision loss benefit from use of a guide dog. A guide dog is a specially trained service animal that helps an individual with vision loss or blindness. Information on guide dog services in the United States can be found at www.guidedogsofamerica.org, and www.guidedog.org.
This resource is funded with support from:
![]()
Prevent Blindness retains full independence over all our health education content, policy positions, advocacy efforts, and programmatic decisions. Find out more about Prevent Blindness and corporate sponsorships.
